Genomics (DNA-Seq)
Whole Genome/Exome Sequencing (WGS/WES)
Whole Genome Sequencing을 통해 원하는 생물종의 전장 유전체를 분석 및 수행합니다. 기본 분석으로 SNP calling, INDEL을 포함한 다양한 영역의 분석 데이터를 제공합니다. 해당 분석은 박테리아부터 식물, 동물세포를 포함하여 Human cell line의 유전체 분석을 포함합니다.
Whole Exome Sequencing은 단백질 코딩 영역만을 집중 분석하여 효율적으로 질환/마커 스페시픽한 주요 유전자 변이를 탐지할 수 있습니다.
Service Features
| Platform (Library) |
Illumina Novaseq (PE150) Nanopore PromethION 48 (8kb) PacBio Revio (15 kb – Hifi) MGI DNBSEQ-T7 (PE150) |
|---|---|
| Sample Type | gDNA |
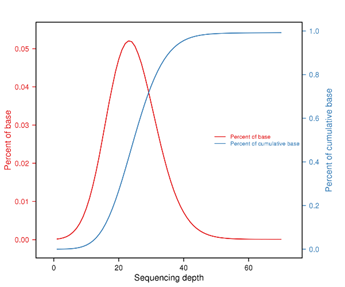
| Sequencing Depth | From 10 – 30 X or more |
| Turnaround Time | ~6 weeks after sample QC |
| Analyzable Species | Bacteria, Yeast, Plant,Animal, Human, Fungi, and other model organisms |
*gDNA prep. 서비스가 필요하신 고객은 상담을 통해 진행 가능합니다.
Service Progress
Data Analysis
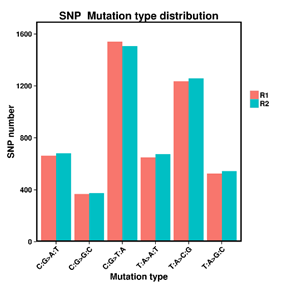
Whole Genome Sequencing의 경우 SNP calling을 통한 Single Nucleotide Polymorphism 분석과 INDEL, CNV (Copy Number Variation) 외 Long Reads Mapping 등 다양한 분석을 고객의 니즈에 맞게 제공합니다.
Whole Exome Sequencing 분석을 통해 단백질 코딩영역 (CDS)만을 집중 분석하여 효율적인 질환 관련 유전자 변이 결과를 제공합니다.
Demo Results
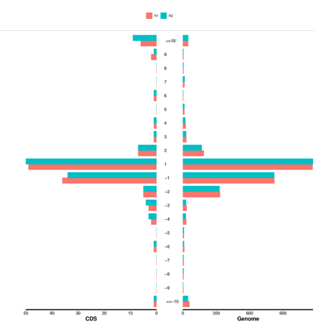
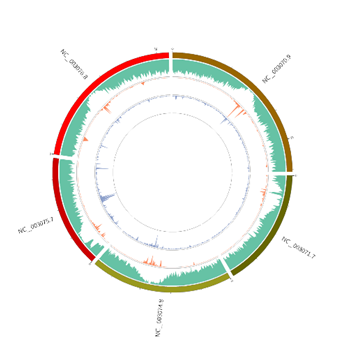
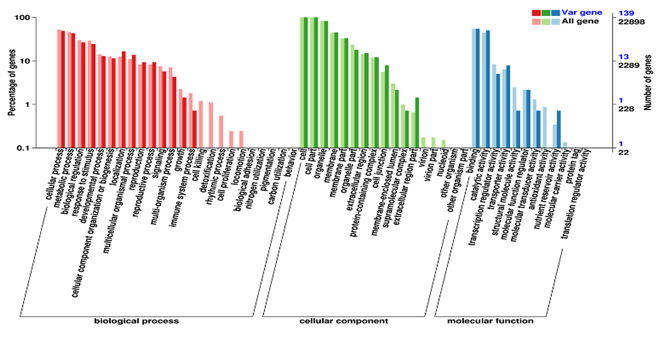
Genomics (DNA-Seq)
De Novo Genome Sequencing & Assembly
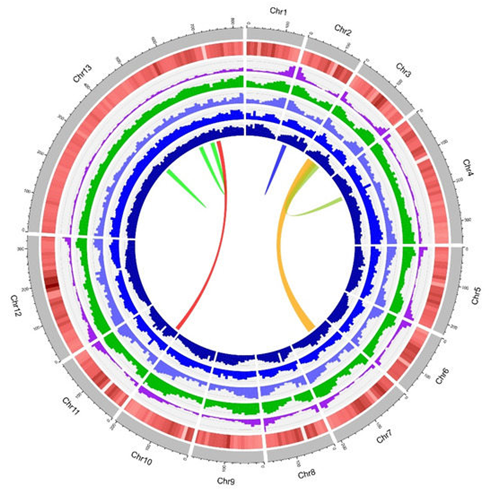
De novo sequencing은 기존의 Reference 유전체 없이, 새로운 생물종의 유전체를 처음부터 조립하는 NGS 분석입니다. 염기 서열 조각들을 중첩 정렬하여 완전한 유전체 서열을 재구성하여 신규 종 해독, 유전체 구조 연구, 비교유전체학 등에 활용됩니다. Long Read Sequence 및 Short Read Sequencing을 통해 정확한 Assembly 서비스를 제공합니다.
Service Features
| Platform (Library) |
Illumina Novaseq (PE150) PacBio CCS HiFi Reads (15 kb – Hifi) |
|---|---|
| Sample Type | gDNA |
| Sequencing Depth | From 10 – 30 X or more |
| Turnaround Time | ~6 weeks after sample QC |
| Analyzable Species | Bacteria, Yeast, Plant,Animal, Human, Fungi, and other model organisms |
*gDNA prep. 서비스가 필요하신 고객은 상담을 통해 진행 가능합니다.
Service Progress
Data Analysis
새로운 종 혹은 미확인 종의 유전체를 처음부터 해독하고 조립하는 De novo Sequencing 분석을 제공합니다. 참조 유전체 없이 고품질의 유전체 구조를 복원하여 유전적 다양성, 진화적 관계, 신규 유전자 탐색 등 다양한 연구에 활용이 가능합니다. 정확한 Assembly 및 Annotation을 연구 목적에 맞게 고객에게 제공합니다.
Demo Results





Genomics (DNA-Seq)
Amplicon Sequencing (16S, 18S, ITS)
16S, 18S, ITS Amplicon sequencing은 미생물 군집의 구성과 다양성을 분석하기 위한 NGS 기반 표준 시퀀싱 서비스입니다. 16S rRNA 영역은 박테리아와 아케이아 동정에, 18S rRNA는 진핵 단세포 생물의 다양성 분석에, ITS 영역은 진균의 종 수준 분류와 생태 연구에 각각 활용됩니다. 이를 통해 다양한 환경, 인체, 동물 시료에서 미생물 군집 구조를 정밀하게 파악할 수 있습니다.
Service Features
| Platform (Library) | PacBio Revio (10/30/50 K Tags) Illumina NovaSeq (PE250, 50/100/300K Tags) |
|---|---|
| Sample Type | Fecal, Soils, Fluids and other Environmental samples |
| Turnaround Time | ~6 weeks after sample QC |
| Analysis Level | Bacteria (Genus to Species), Fungi, and other eukaryotes |
*gDNA prep. 서비스가 필요하신 고객은 상담을 통해 진행 가능합니다.
Service Progress
Data Analysis
대변을 포함한 다양한 샘플 내 존재하는 박테리아 혹은 진균들의 분포를 확인하는 서비스입니다. 16S 분석의 경우에는 박테리아를 주요 타겟으로 하여 V3-V4 영역을 증폭하는 방식과 전체 길이의 16S 시퀀스를 분석하여 Species level까지 동정하실 수 있는 서비스를 제공합니다. 마찬가지로 ITS 및 18S 영역 분석 서비스를 제공하여 고객의 니즈에 맞는 Amplicon 서비스를 제공합니다.
Demo Results





Genomics (DNA-Seq)
Metagenome Sequencing
Metagenome sequencing (메타게놈 시퀀싱) 서비스는 토양, 대변, 해수를 포함하는 다양한 환경 시료로부터 전체 미생물 군집의 DNA를 추출하여 분석하는 기술로, 박테리아 어려운 미생물을 포함한 미생물의 구성 및 기능적 잠재력을 볼 수 있는 서비스입니다. 유전자 레벨의 분석을 통해 단백질 구성의 기능을 유추할 수 있습니다.
Service Features
| Platform (Library) |
Illumina NovaSeq (PE150, 6GB) Nanopore P48 (10kb, 6/10GB) PacBio Revio (8kb, 10/20GB) |
|---|---|
| Sample Type | Fecal, Soils, Fluids and other Environmental samples |
| Turnaround Time | ~6 weeks after sample QC |
| Analysis Level | Compositional and Functional analysis |
*gDNA prep. 서비스가 필요하신 고객은 상담을 통해 진행 가능합니다.
Service Progress
Data Analysis
추출된 Total DNA를 라이브러리로 제작한 뒤 고객의 니즈에 알맞은 시퀀싱 플랫폼으로 분석을 수행합니다.
이후 생물정보학 분석을 통해 유전체 조립, Annotation, 분류학적 구성과 기능적 프로파일링 결과를 제공합니다.
Demo Results









Genomics (DNA-Seq)
Specific-Locus Amplified Fragment Sequencing (SLAF-Seq)
SLAF 시퀀싱 기술은 제한효소를 이용해 유전체를 효율적으로 절단한 후 선택된 특정 길이의 DNA 단편을 선택적으로 시퀀싱하여
유전체 전반의 변이(SNP)들을 빠르게 탐색하는 기술입니다.
d전장 유전체 시퀀싱 대비 고밀도의 마커정보를 얻을 수 있어, Comparative study 혹은 형질연관분석 (GWAS) 등에 널리 활용될 수 있습니다.
Service Features
| Platform (Library) |
Illumina NovaSeq (PE150) Processed with Restriction Enzymes |
|---|---|
| Sample Type | Samples, gDNA |
| Turnaround Time | ~6 weeks after sample QC |
| Analysis Type |
Genetic Maps [Parents: 20X WGS, Offspring: 10X] Genome-Wide Association Studies (GWAS) [over 200 samples, 10X] Genetic Evolution [over 30 samples, w/ over 10 samples from each, 10X] |
*gDNA prep. 서비스가 필요하신 고객은 상담을 통해 진행 가능합니다.
Service Progress







Data Analysis
고객의 샘플 및 DNA를 활용하여 in silico design부터 제한효소 선택까지 원스텝 서비스를 제공합니다. 이후 시퀀싱 및 SLAF 시퀀싱 서비스를 활용해 SNP 분석 및 Genome clustering 등 다양한 서비스를 고객 니즈에 맞게 제공합니다.
High Genetic Marker Discovery: We integrate a high-throughput double barcode system allowing for the simultaneous sequencing of large populations, and locus-specific amplification enhancing efficiency, ensuring that tag numbers meet the diverse requirements of various research questions.
Low Dependence on the Genome: It can be applied to species with or without a reference genome.
Flexible Scheme Design: Single-enzyme, dual-enzyme, multi-enzyme digestion, and various types of enzymes can all be selected to cater to different research goals or species.
High Efficiency In Enzymatic Digestion: The conduction of an in-silico pre-design and a pre-experiment assures optimal design with even distribution of SLAF tags on the chromosome (1 SLAF tag/4Kb) and reduced repetitive sequence (<5%).
Extensive Expertise: We bring a wealth of experience to every project, with a track record of closing over 5000 SLAF-Seq projects on hundreds of species, including plants, mammals, birds, insects, and aquatic organisms.
Self-developed Bioinformatic Workflow: We developed an integrated bioinformatic workflow for SLAF-Seq to ensure the reliability and accuracy of the final output.
Demo Results




Genomics (DNA-Seq)
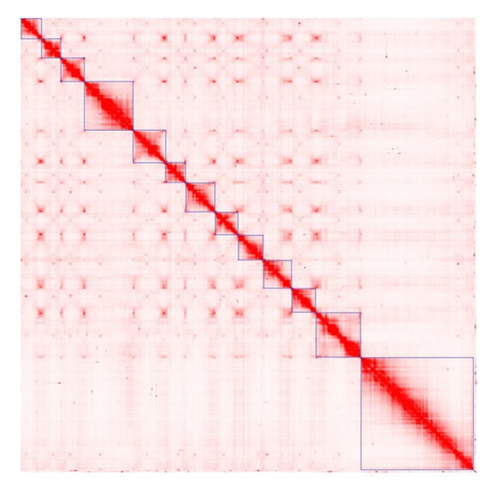
Hi-C Based Genome Assembly
Hi-C Based Genome Assembly는 Chromosome 수준에서 유전체를 정교하게 재구성하기 위한 기술로, 세포 내에서 공간적으로 가까이 존재하는 DNA Fragment들의 교차결합 특성을 활용합니다. Hi-C를 활용한 기술과 기존의 Short-Read 혹은 Long-Read 시퀀스를 통합하면 동일 염색체 조각들을 정확한 순서와 방향으로 연결할 수 있어 Contig 수준의 결과물을 Scaffold 수준으로 확장시킬 수 있습니다. 또한 복합 염색체 수준의 고해상도 Assembly가 가능해져 복잡한 유전체를 가진 생물 및 반복서열을 가지는 종에서도 매우 유용한 기술입니다.
Service Features
| Platform (Library) | Hi-C Library (Illumina NovaSeq PE150, 100X) |
|---|---|
| Sample Type | Sample only (Such as Tissues and Cells) |
| Turnaround Time | ~6 weeks after sample QC |
| Applicable samples | Animal Viscera, Muscles, Mammalian Blood, Poultry/Fish Blood, Plant, Cultured Cells, and Insects |
Service Progress



• Sequencing on Illumina NovaSeq with PE150.
• Service requires tissue samples, instead of extracted nucleic acids, to cross-link with formaldehyde and conserve the DNA-protein interactions.
• The Hi-C experiment involves restriction and end repair of the sticky ends with biotin, followed by circularization of the resulting blunt ends while preserving the interactions. The DNA is then pulled down with streptavidin beads and purified for subsequent library preparation.
Data Analysis
Hi-C Based Genome Assembly는 염색체 간 3차원 상호작용 정보를 활용하여 염색체 수준의 정밀한 유전체 구조를 재구성합니다. 기존 시퀀싱 데이터와 통합하여 정확한 Scaffold 영역과 High-Quality Genome Assembly Service를 고객의 니즈에 맞게 제공합니다.
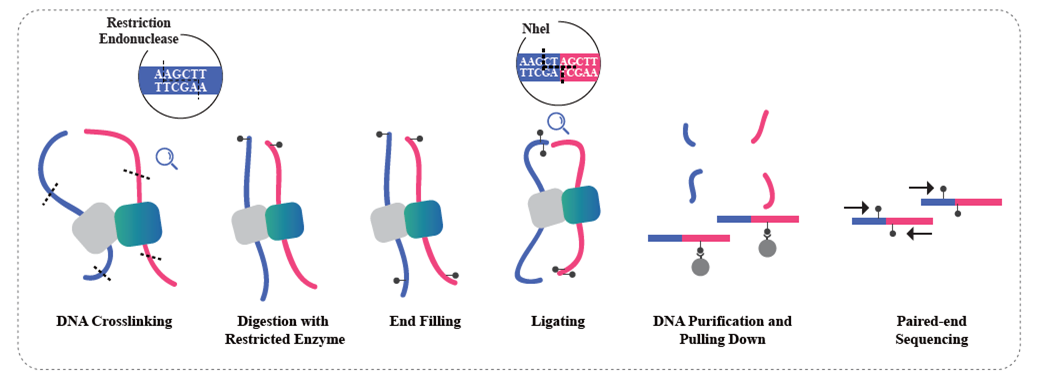
Genetic Population Data Free
The Hi-C technology substitutes the essential information required for contig anchoring, enabling highly accurate genome assembly without the need for additional genetic population data.
High Marker Density
Achieves a high contig anchoring rate exceeding 90%, ensuring robust and precise genome scaffolding.
Extensive Expertise and Publications
With experience in over 2,000 Hi-C–based genome assemblies across more than 1,000 species, our company possesses numerous patents. Over 200 of our studies have been published with a cumulative impact factor exceeding 2,000.
Highly Skilled Bioinformatics Team
We own proprietary software copyrights and patents for Hi-C experimental and data analysis tools, enabling efficient data processing and editing through our self-developed visualization platforms.
Post-Sales Support
We provide three months of post-project support, during which our team addresses inquiries, resolves issues, and conducts Q&A sessions to ensure complete customer satisfaction.
Comprehensive Annotation
By integrating multiple databases, we perform functional annotation of identified genetic variants and conduct enrichment analyses to deliver comprehensive, project-tailored insights that meet diverse research needs.
Demo Results